Фенилкетонурия (болезнь Феллинга) - методы лечения и другие ответы от эксперта Эко-Блог
Автор статьи:

Статья проверена:
Барахоева Зарема Бекхановна , репродуктолог по лечению бесплодия
Что это такое
Фенилкетонурия возникает при нарушении обмена веществ в результате дефицита одного из ферментов фенилаланина, вследствие чего активируются побочные пути обмена, что вызывает накопление токсичных продуктов, которых в норме в организме человека нет. Часть из них провоцирует нарушения липидного обмена в головном мозге, приводящие к поражению центральной нервной системы.
Причины фенилкетонурии
При классической форме заболевания, так называемой фенилкетонурии I типа, встречающейся в 98% всех диагностированных случаев, в основе лежит недостаток печеночного фермента, который поступает с пищей и участвует в превращении фенилаланина в тирозин (входит в состав пигмента, ферментов, необходим для функционирования всего организма). В результате концентрация фенилаланина в крови, спинномозговой жидкости увеличивается, соответственно, концентрация тирозина падает. Процессу сопутствует нарушение миелинизации нервных волокон, что ведет к уменьшению образования нейромедиаторов, которые участвуют в передаче нервных импульсов между клетками нервной системы, в результате запускается механизм отставания умственного развития.

Помимо классического типа, существуют атипичные формы болезни, причины фенилкетонурии в этом случае связаны с мутацией в других генах, которые ответственны за кодирование фермента. Вероятность рождения ребенка с подобной патологией у родителей-носителей мутантного гена составляет 1:4. Клинические проявления подобны симптомам при классической форме болезни, однако данная патология не поддается коррекции с помощью диеты.
Следует заметить, что риск развития патологии увеличивается при рождении ребенка у близких родственников. Для появления симптоматики ребенок должен получить от обоих родителей (носителей мутантного гена) по одной дефектной копии гена; такая вероятность в случае брака между ближайшими родственниками существенно возрастает. В этом случае заболеванию сопутствует глубокая умственная отсталость.
Симптомы

С момента рождения ребенок выглядит абсолютно здоровым, поэтому важно с первых дней жизни выявить недуг. Первые симптомы фенилкетонурии возникают у ребенка в первом полугодии. При поступлении в организм малыша грудного молока или его заменителей развиваются начальные признаки: вялость, беспокойство, ребенок мало двигается, не исключено появление судорог, упорной рвоты, возможна гипервозбудимость. Слабо реагируя на происходящее, ребенок не узнает мать. По достижении шести месяцев становится заметным отставанием в психическом развитии.
У детей с такой патологией поздно появляются зубы, сидеть и ходить они начинают гораздо позже своих сверстников. В год малыши не понимают речь родителей, не способны выразить эмоции голосом. Фенилаланин выводится из организма с мочой и потом, поэтому от пациента исходит специфический затхлый запах, напоминающий мышиный.
Позже появляется характерное положение тела: ноги широко расставлены, согнуты в коленных и тазобедренных суставах, плечи и голова опущены. Походка мелкими шагами, с небольшим покачиванием. Из-за повышенного мышечного тонуса сидит такой пациент в позе портного — поджав ноги. Отмечается недостаток пигмента, у больного малыша светлые глаза и гипопигментированные волосы. Нередко возникает экзема, дерматиты, катаракта. К четырем годам у такого ребенка речь полностью отсутствует. Без проводимого лечения возникает микроцефалия, тремор рук, шаткая походка, акроцианоз (синюшность кожи), прогнатия (аномалия прикуса), гиперкинезы (непроизвольные движения). Также возможна дисфункция печени.
Осложнения фенилкетонурии
Прогрессирование недуга приводит к гибели пациента в двух-трехлетнем возрасте. Кроме того, частое осложнение фенилкетонурии — эпилептические приступы. Возникающие на 2-ом году жизни ребенка, они плохо купируются противосудорожными препаратами.
Диагностика
Очень важно распознать болезнь в доклинической стадии, а при появлении начальных признаков в первые два месяца жизни ребенка срочно начать лечение. Поэтому на 3-4 сутки после появления малыша на свет проводится неонатальный скрининг для диагностики фенилкетонурии. Для подтверждения диагноза с помощью исследования крови определяется уровень фенилаланина и тирозина, активность печеночных ферментов, по достижении 10-12 дня жизни ребенка проводится биохимическое исследование мочи. Также выполняется ЭЭГ и МРТ головного мозга.
Генетический дефект у плода можно выявить в помощью пренатальной диагностики в период внутриутробного развития, поэтому при существующей вероятности развития недуга беременным рекомендуется пройти консультацию у специалиста.
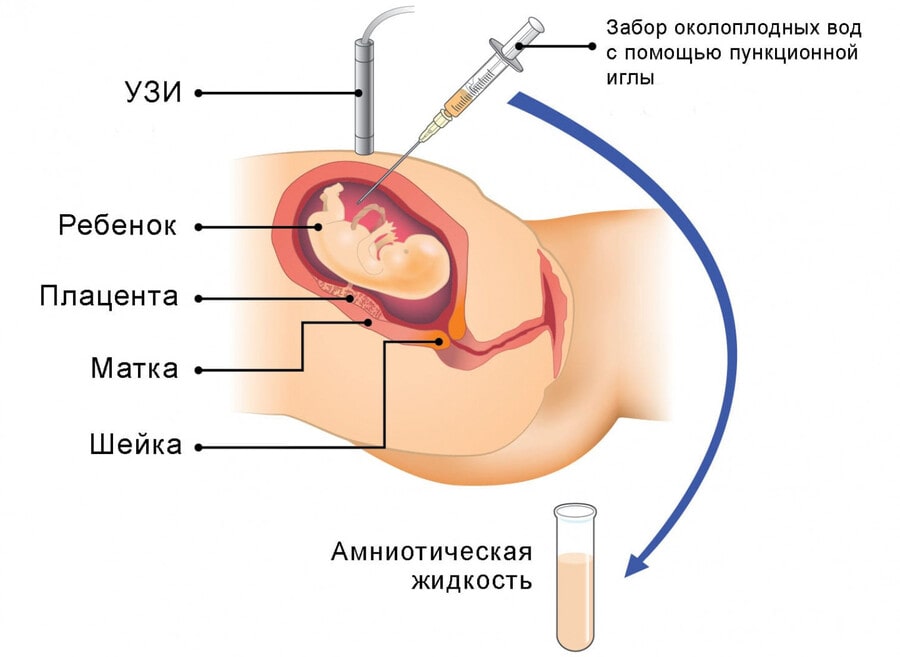
Как лечить фенилкетонурию
К сожалению, лекарственных средств, которые позволили бы контролировать показатели аминокислоты без соблюдения диеты, пока нет. Атипичные формы болезни корректируются с помощью препаратов тетрагидробиоптерина и его аналогов. Для заболевания I типа единственным действенным методом лечения фенилкетонурии остается диетотерапия. Ее принцип основан на ограничении приема фенилаланина, содержащегося в продуктах. Хлеб, орехи, бобовые, рыба, мясо, творог, крупы, яйца, шоколад и др. следует исключить из рациона. Для детей первого года жизни и тем, кто немного постарше, разработаны специальные смеси, максимально приближенные к грудному молоку. Также существуют варианты для беременных и детей, достигших 6-летнего возраста. Если ограничивать поступление в организм фенилаланина с пищей до полового созревания, то патологических изменений можно избежать. Однако следует учитывать, что при отсутствии лечения резвившиеся нарушения в тканях мозга являются необратимыми, поэтому лечебная диета должна применяться с самого рождения.
Для восполнения дефицита белка, витаминов и микроэлементов, который неминуем при ограничительной диете в течение длительного времени, необходимо компенсировать недостаток специальными препаратами (белковые гидролизаты, смеси аминокислот, витамины, препараты железа и т.д.). Кроме того, в медикаментозное лечение входит прием препаратов, которые улучшают сосудистую микроциркуляцию. В зависимости от состояния здоровья назначаются антиконвульсанты.
Дети с фенилкетонурией должны находится на постоянном учете у педиатра и психоневролога. Нередко пациенты нуждаются в помощи логопеда. Поначалу контроль показателей проводится каждую неделю, затем, после нормализации состояния, ребенок до года обследуется один раз в месяц, дети более старшего возраста должны посещать врача раз в два месяца.
Профилактика
Для того, чтобы оценить вероятность рождения больного ребенка, супруги, являющиеся близкими родственниками, должны пройти консультацию у генетика. Женщинам, у которых уже есть больной ребенок с диагнозом фенилкетонурия, необходимо при планируемом рождении предпринять профилактические меры задолго до зачатия и во время самой беременности. Своевременно начатая диетотерапия позволит предупредить нарушение развития плода.
Список использованной литературы:
- Бушуева Т.В., Боровик Т.Э., Ладодо К.С., Кузен-кова Л.М., Маслова О.И., Геворкян А.К. Оценка физического развития у детей с классической фенилкетонурией. Вопр. пит. 2015; 84 (2): 34—43.
- Бушуева Т.В., Винярская И.В., Черников В.В., Боровик Т.Э., Кузенкова Л.М. Оценка качества жизни детей, больных фенилкетонурией. Вестник Российской академии медицинских наук 2014; 11—12: 39—45.
- Врожденные заболевания. В кн.: Руководство по педиатрии / Под ред. А. А. Баранова, Б. С. Каганова, Р Р. Шиляева. М., 2007. 544 с.
- Наследственные нарушения нервно-психического развития детей / Под ред. А. Темина, Л. З. Казанцевой. М., 2001. 429 с.
- Баранов А. А., Боровик Т. Э., Ладодо К. С. и соавт. Новые специализированные лечебные продукты для питания детей, больных фенилкетонурией (пособие для врачей). М.: МЗ СР РФ, 2005. 88 с.
Консультации специалистов
Генетика
| Наименование услуги | Стоимость, руб. |
| Первичная консультация генетика | 2 700 |
| Повторная консультация генетика | 2 500 |
| Консультация генетика по скайпу или телефону | 2 500 |
Эндокринология
| Наименование услуги | Стоимость, руб. |
| Консультация эндокринолога | 2 300 |








